Zucco, Adrian G.
Exploring Nationwide Patterns of Sleep Problems from Late Adolescence to Adulthood Using Machine Learning
Zucco, Adrian G.
, Drews, Henning Johannes
, Uleman, Jeroen F.
, Bhatt, Samir
, Rod, Naja Hulvej
Sleep
Pattern Recognition
Life Course
Complexity
Sleep problems among young adults pose a major public health challenge. Leveraging nationwide health surveys and registers from Denmark, we investigated patterns of sleep problems from late adolescence to adulthood and explored early life-course determinants. We generated life-course embeddings using unsupervised machine learning on data from 2.2 million individuals born from 1980 to 2015. We used this landscape to identify neighboring factors of sleep problems. We observed a substantial increase in self-reported sleep problems among individuals aged 15 to 45, from 34 to 49% between 2010 and 2021, and a 10-fold increase in melatonin use. We also found relevant clusters of sleep-related prescriptions, diagnoses, and procedures with age-specific incidence patterns. Specific childhood adversities, such as sibling psychiatric illness, foster care, and parental divorce, were shared factors across multiple sleep disorders such as insomnia and nightmares. These findings underscore the complex interplay between medical and psychosocial factors in sleep.
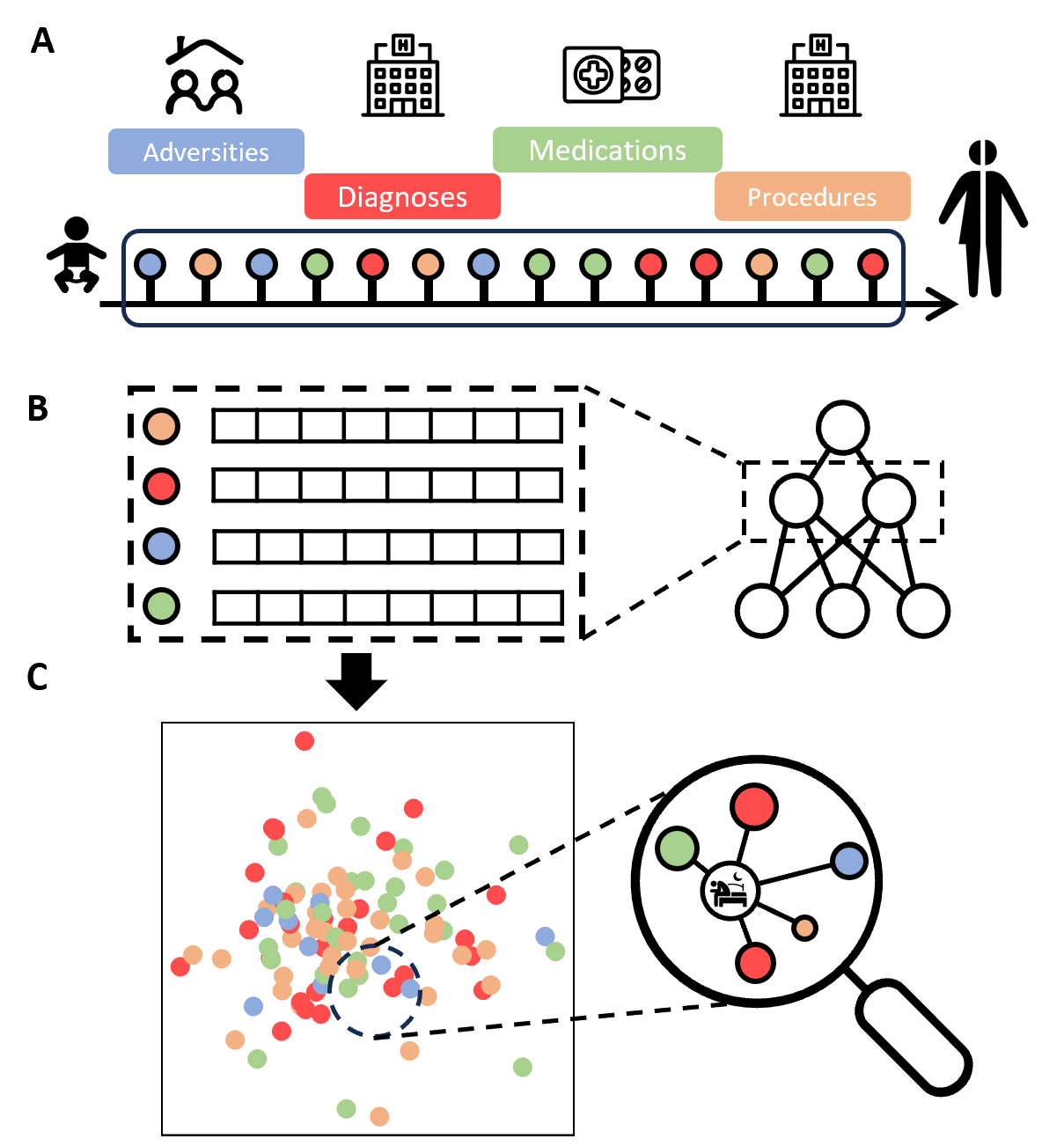
Metabolic Profiling Early Post-Allogeneic Haematopoietic Cell Transplantation in the Context of CMV Infection
Rasmussen, Kirstine K.
, Dos Santos, Quenia
, MacPherson, Cameron Ross
, Zucco, Adrian G.
, Gjærde, Lars Klingen
, Ilett, Emma E.
, Lodding, Isabelle
, Helleberg, Marie
, Lundgren, Jens D.
, Nielsen, Susanne D.
, Brix, Susanne
, Sengeløv, Henrik
, Murray, Daniel D.
AHSCT
CMV
Correlation Network Analysis
Cytomegalovirus
Lipidomics
Metabolomics
TMAO
WGCNA
Immune dysfunction resulting from allogeneic haematopoietic stem cell transplantation (aHSCT) predisposes one to an elevated risk of cytomegalovirus (CMV) infection. Changes in metabolism have been associated with adverse outcomes, and in this study, we explored the associations between metabolic profiles and post-transplantation CMV infection using plasma samples collected 7–33 days after aHSCT. We included 68 aHSCT recipients from Rigshospitalet, Denmark, 50% of whom experienced CMV infection between days 34–100 post-transplantation. First, we investigated whether 12 metabolites selected based on the literature were associated with an increased risk of post-transplantation CMV infection. Second, we conducted an exploratory network-based analysis of the complete metabolic and lipidomic profiles in relation to clinical phenotypes and biological pathways. Lower levels of trimethylamine N-oxide were associated with subsequent CMV infection (multivariable logistic regression: OR = 0.63; 95% CI = [0.41; 0.87]; p = 0.01). Explorative analysis revealed 12 clusters of metabolites or lipids, among which one was predictive of CMV infection, and the others were associated with conditioning regimens, age upon aHSCT, CMV serostatus, and/or sex. Our results provide evidence for an association between the metabolome and CMV infection post-aHSCT that is independent of known risk factors.
Associations of Functional Human Leucocyte Antigen Class I Groups with HIV Viral Load in a Heterogeneous Cohort
Zucco, Adrian G.
, Bennedbæk, Marc
, Ekenberg, Christina
, Gabrielaite, Migle
, Leung, Preston
, Polizzotto, Mark N.
, Kan, Virginia
, Murray, Daniel D.
, Lundgren, Jens D.
, MacPherson, Cameron R.
Objective:~ Human leucocyte antigen (HLA) class I alleles are the main host genetic factors involved in controlling HIV-1 viral load (VL). Nevertheless, HLA diversity has proven a significant challenge in association studies. We assessed how accounting for binding affinities of HLA class I alleles to HIV-1 peptides facilitate association testing of HLA with HIV-1 VL in a heterogeneous cohort. Design:~ Cohort from the Strategic Timing of AntiRetroviral Treatment (START) study. Methods:~ We imputed HLA class I alleles from host genetic data (2546 HIV+ participants) and sampled immunopeptidomes from 2079 host-paired viral genomes (targeted amplicon sequencing). We predicted HLA class I binding affinities to HIV-1 and unspecific peptides, grouping alleles into functional clusters through consensus clustering. These functional HLA class I clusters were used to test associations with HIV VL. Results:~ We identified four clades totaling 30 HLA alleles accounting for 11.4% variability in VL. We highlight HLA-B$$57:01 and B$$57:03 as functionally similar but yet overrepresented in distinct ethnic groups, showing when combined a protective association with HIV+ VL (log, $β$ -0.25; adj. P-value $<$ 0.05). We further demonstrate only a slight power reduction when using unspecific immunopeptidomes, facilitating the use of the inferred functional HLA groups in other studies Conclusion:~ The outlined computational approach provides a robust and efficient way to incorporate HLA function and peptide diversity, aiding clinical association studies in heterogeneous cohorts. To facilitate access to the proposed methods and results we provide an interactive application for exploring data.
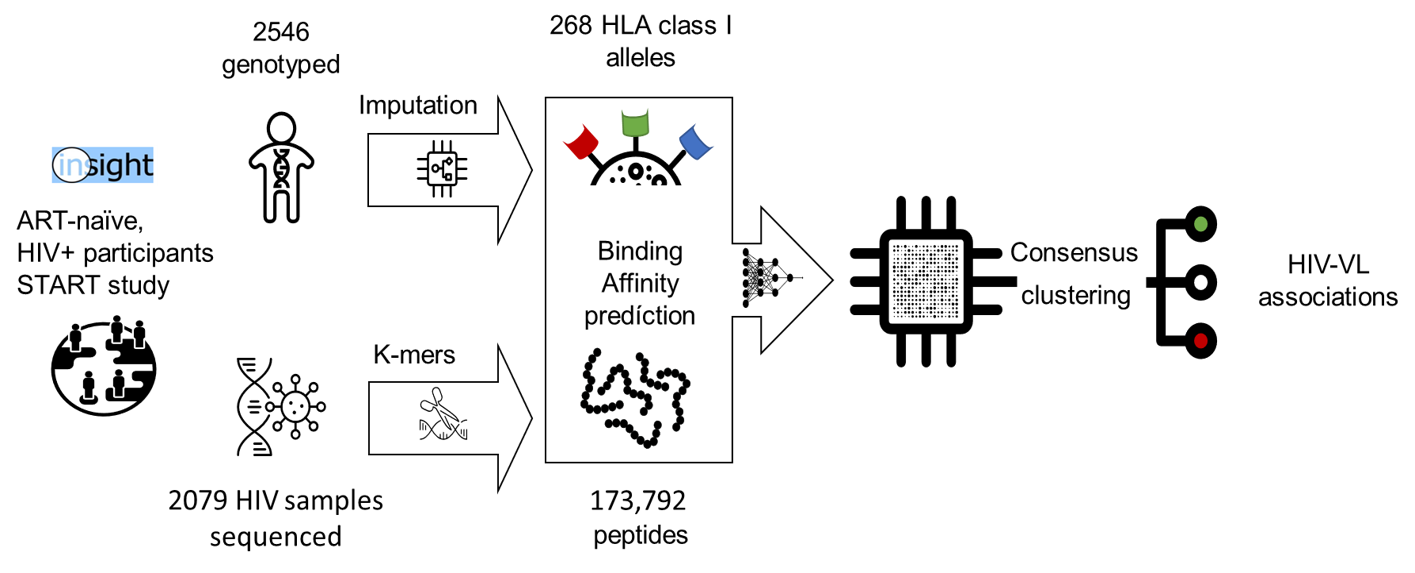
Association between Ten-Eleven Methylcytosine Dioxygenase 2 Genetic Variation and Viral Load in People with HIV
Murray, Daniel D.
, Grund, Birgit
, MacPherson, Cameron R.
, Ekenberg, Christina
, Zucco, Adrian G.
, Reekie, Joanne
, Dominguez-Dominguez, Lourdes
, Leung, Preston
, Fusco, Dahlene
, Gras, Julien
, Gerstoft, Jan
, Helleberg, Marie
, Borges, Álvaro H.
, Polizzotto, Mark N.
, Lundgren, Jens D.
Introduction:~ Identifying genetic factors that influence HIV-pathogenesis is critical for understanding disease pathways. Previous studies have suggested a role for the human gene ten-eleven methylcytosine dioxygenase 2 (TET2) in modulating HIV-pathogenesis. Methods:~ We assessed whether genetic variation in TET2 was associated with markers of HIV-pathogenesis using both gene level and single nucleotide polymorphism (SNP) level association in 8512 HIV-positive persons across five clinical trial cohorts. Results:~ Variation at both the gene and SNP-level of TET2 was found to be associated with levels of HIV viral load (HIV-VL) consistently in the two cohorts that recruited antiretroviral-naïve participants. The SNPs occurred in two clusters of high linkage disequilibrium (LD), one associated with high HIV-VL and the other low HIV-VL, and were predominantly found in Black participants. Conclusion:~ Genetic variation in TET2 was associated with HIV-VL in two large antiretroviral therapy (ART)-naive clinical trial cohorts. The role of TET2 in HIV-pathogenesis warrants further investigation.
Chest X-Ray Imaging Score Is Associated with Severity of COVID-19 Pneumonia: The MBrixia Score
Jensen, Christian M.
, Costa, Junia C.
, Nørgaard, Jens C.
, Zucco, Adrian G.
, Neesgaard, Bastian
, Niemann, Carsten U.
, Ostrowski, Sisse R.
, Reekie, Joanne
, Holten, Birgit
, Kalhauge, Anna
, Matthay, Michael A.
, Lundgren, Jens D.
, Helleberg, Marie
, Moestrup, Kasper S.
Respiratory Signs and Symptoms
Viral Infection
Spatial resolution in existing chest x-ray (CXR)-based scoring systems for coronavirus disease 2019 (COVID-19) pneumonia is low, and should be increased for better representation of anatomy, and severity of lung involvement. An existing CXR-based system, the Brixia score, was modified to increase the spatial resolution, creating the MBrixia score. The MBrixia score is the sum, of a rule-based quantification of CXR severity on a scale of 0 to 3 in 12 anatomical zones in the lungs. The MBrixia score was applied to CXR images from COVID-19 patients at a single tertiary hospital in the period May 4th–June 5th, 2020. The relationship between MBrixia score, and level of respiratory support at the time of performed CXR imaging was investigated. 37 hospitalized COVID-19 patients with 290 CXRs were identified, 22 (59.5%) were admitted to the intensive care unit and 10 (27%) died during follow-up. In a Poisson regression using all 290 MBrixia scored CXRs, a higher MBrixia score was associated with a higher level of respiratory support at the time of performed CXR. The MBrixia score could potentially be valuable as a quantitative surrogate measurement of COVID-19 pneumonia severity, and future studies should investigate the score’s validity and capabilities of predicting clinical outcomes.
Personalized Survival Probabilities for SARS-CoV-2 Positive Patients by Explainable Machine Learning
Zucco, Adrian G.
, Agius, Rudi
, Svanberg, Rebecka
, Moestrup, Kasper S.
, Marandi, Ramtin Z.
, MacPherson, Cameron Ross
, Lundgren, Jens
, Ostrowski, Sisse R.
, Niemann, Carsten U.
Machine Learning
Prognosis
Viral Infection
Interpretable risk assessment of SARS-CoV-2 positive patients can aid clinicians to implement precision medicine. Here we trained a machine learning model to predict mortality within 12~weeks of a first positive SARS-CoV-2 test. By leveraging data on 33,938 confirmed SARS-CoV-2 cases in eastern Denmark, we considered 2723 variables extracted from electronic health records (EHR) including demographics, diagnoses, medications, laboratory test results and vital parameters. A discrete-time framework for survival modelling enabled us to predict personalized survival curves and explain individual risk factors. Performance on the test set was measured with a weighted concordance index of 0.95 and an area under the curve for precision-recall of 0.71. Age, sex, number of medications, previous hospitalizations and lymphocyte counts were identified as top mortality risk factors. Our explainable survival model developed on EHR data also revealed temporal dynamics of the 22 selected risk factors. Upon further validation, this model may allow direct reporting of personalized survival probabilities in routine care.
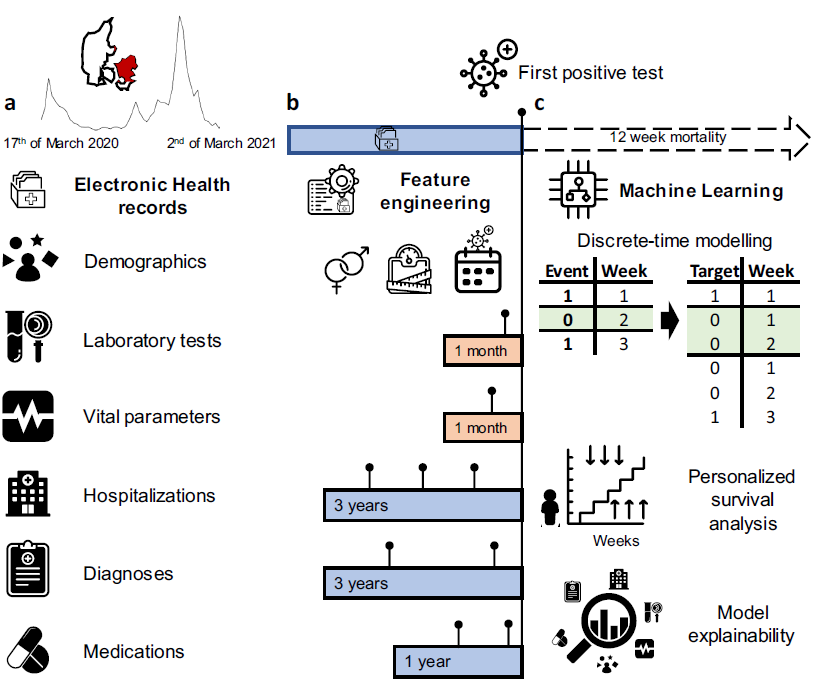
Explainable Machine Learning for Precision Medicine of Patients with Infectious Diseases
Zucco, Adrian G.
This thesis aims to present Machine Learning applications for the development of precision medicine in patients with infectious diseases. This is outlined by proposing computational solutions to two major challenges in precision medicine: how to infer relevant host genetic factors in heterogeneous populations (Study I) and how to predict patient-specific risk while accounting for censored individuals (Study II). For both challenges, the implemented models are explained based on domain knowledge of biological systems and disease aetiology supported by methods of model interpretability. This corresponds to the secondary aim of developing not only predictive models but also deepening the understanding of HIV host genomics and SARS-CoV-2 risk factors respectively. The specific objectives of each study were: Study I – Associations of functional HLA class I groups with HIV viral load in a heterogeneous cohort. To assess if functional clustering of the main host genetic factors involved in HIV control, Human Leukocyte Antigen alleles, based on predicted binding affinities to HIV peptides facilitate the study of HLA alleles in demographically heterogeneous cohorts. Study II – Personalized survival probabilities for SARS-CoV-2 positive patients by explainable machine learning. To implement survival machine learning models for predicting personalized 12-week mortality of SARS-CoV-2 positive patients by leveraging electronic health records and describing temporal dynamics of relevant risk factors through model explainability.

The Association of Human Leukocyte Antigen Alleles with Clinical Disease Progression in HIV-positive Cohorts with Varied Treatment Strategies
Ekenberg, Christina
, Reekie, Joanne
, Zucco, Adrian G.
, Murray, Daniel D.
, Sharma, Shweta
, MacPherson, Cameron R.
, Babiker, Abdel
, Kan, Virginia
, Lane, H. Clifford
, Neaton, James D.
, Lundgren, Jens D.
, For the INSIGHT START, SMART Study Groups
Objectives:~ The Strategic Timing of AntiRetroviral Treatment (START) and Strategies for Management of Antiretroviral Therapy (SMART) trials demonstrated that ART can partly reverse clinically defined immune dysfunction induced by HIV replication. As control of HIV replication is influenced by the HLA region, we explored whether HLA alleles independently influence the risk of clinical events in HIV+ individuals. Design:~ Cohort study. Methods:~ In START and SMART participants, associations between imputed HLA alleles and AIDS, infection-related cancer, herpes virus-related AIDS events, chronic inflammation-related conditions, and bacterial pneumonia were assessed. Cox regression was used to estimate hazard ratios for the risk of events among allele carriers versus noncarriers. Models were adjusted for sex, age, geography, race, time-updated CD4+ T-cell counts and HIV viral load and stratified by treatment group within trials. HLA class I and II alleles were analyzed separately. The Benjamini–Hochberg procedure was used to limit the false discovery rate to less than 5% (i.e. q value $<$0.05). Results:~ Among 4829 participants, there were 132 AIDS events, 136 chronic inflammation-related conditions, 167 bacterial pneumonias, 45 infection-related cancers, and 49 herpes virus-related AIDS events. Several associations with q value less than 0.05 were found: HLA-DQB1$$06:04 and HLA-DRB1$$13:02 with AIDS (adjusted HR [95% CI] 2.63 [1.5–4.6] and 2.25 [1.4–3.7], respectively), HLA-B$$15:17 and HLA-DPB1$$15:01 with bacterial pneumonia (4.93 [2.3–10.7] and 4.33 [2.0–9.3], respectively), and HLA-A$*$69:01 with infection-related cancer (15.26 [3.5–66.7]). The carriage frequencies of these alleles were 10% or less. Conclusion:~ This hypothesis-generating study suggests that certain HLA alleles may influence the risk of immune dysfunction-related events irrespective of viral load and CD4+ T-cell count.
Association Between Single-Nucleotide Polymorphisms in HLA Alleles and Human Immunodeficiency Virus Type 1 Viral Load in Demographically Diverse, Antiretroviral Therapy--Naive Participants From the Strategic Timing of AntiRetroviral Treatment Trial
Ekenberg, Christina
, Tang, Man-Hung
, Zucco, Adrian G.
, Murray, Daniel D.
, MacPherson, Cameron Ross
, Hu, Xiaojun
, Sherman, Brad T.
, Losso, Marcelo H.
, Wood, Robin
, Paredes, Roger
, Molina, Jean-Michel
, Helleberg, Marie
, Jina, Nureen
, Kityo, Cissy M.
, Florence, Eric
, Polizzotto, Mark N.
, Neaton, James D.
, Lane, H. Clifford
, Lundgren, Jens D.
``To investigate the impact of host genetics on human immunodeficiency virus type 1 control among individuals of different ancestry, we performed genome-wide ass